ParDOCK
ParDOCK is an all-atom energy based Monte Carlo,rigid protein ligand docking, implemented in a fully automated, parallel processing mode which predicts the binding mode of the ligand in receptor target site[1]
The structural input data for the ParDOCK are optimized reference complex i.e. protein bound with a ligand (Sample File) and a candidate ligand to be docked(Sample file).
ParDOCK has been tested on 226 protein-ligand complexes.
Click here to access the 226 protein ligand dataset.
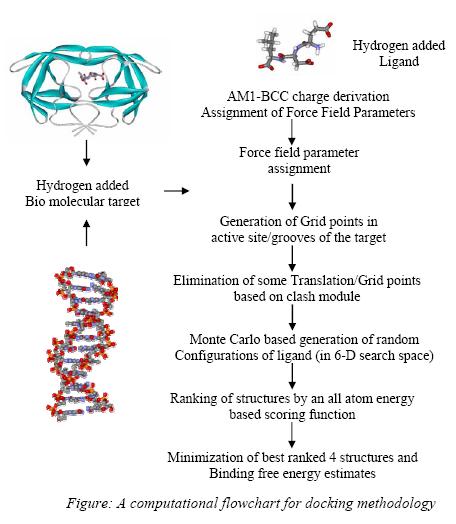
[1] Gupta, A. Gandhimathi, A. Sharma, P. and Jayaram, B. (2007) ParDOCK: An All Atom Energy Based Monte Carlo Docking Protocol for Protein-Ligand Complexes. Protein and Peptide Letters, 2007, 14, 7, 632-646.