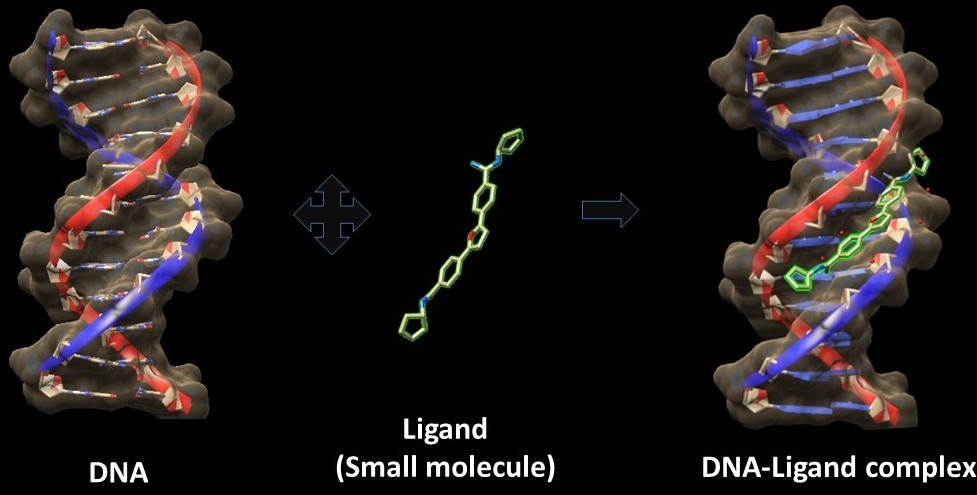
DNA Ligand Docking is an all-atom energy based Monte Carlo DNA ligand docking,
implemented in a fully automated, parallel processing mode which predicts the
binding mode of the non-metallo ligand in the minor groove of DNA. The input is a DNA sequence and drug PDB file. The output will a docked structure alongwith the binding affinity of the docked structures.
'How to Use Tool'.
| 