About
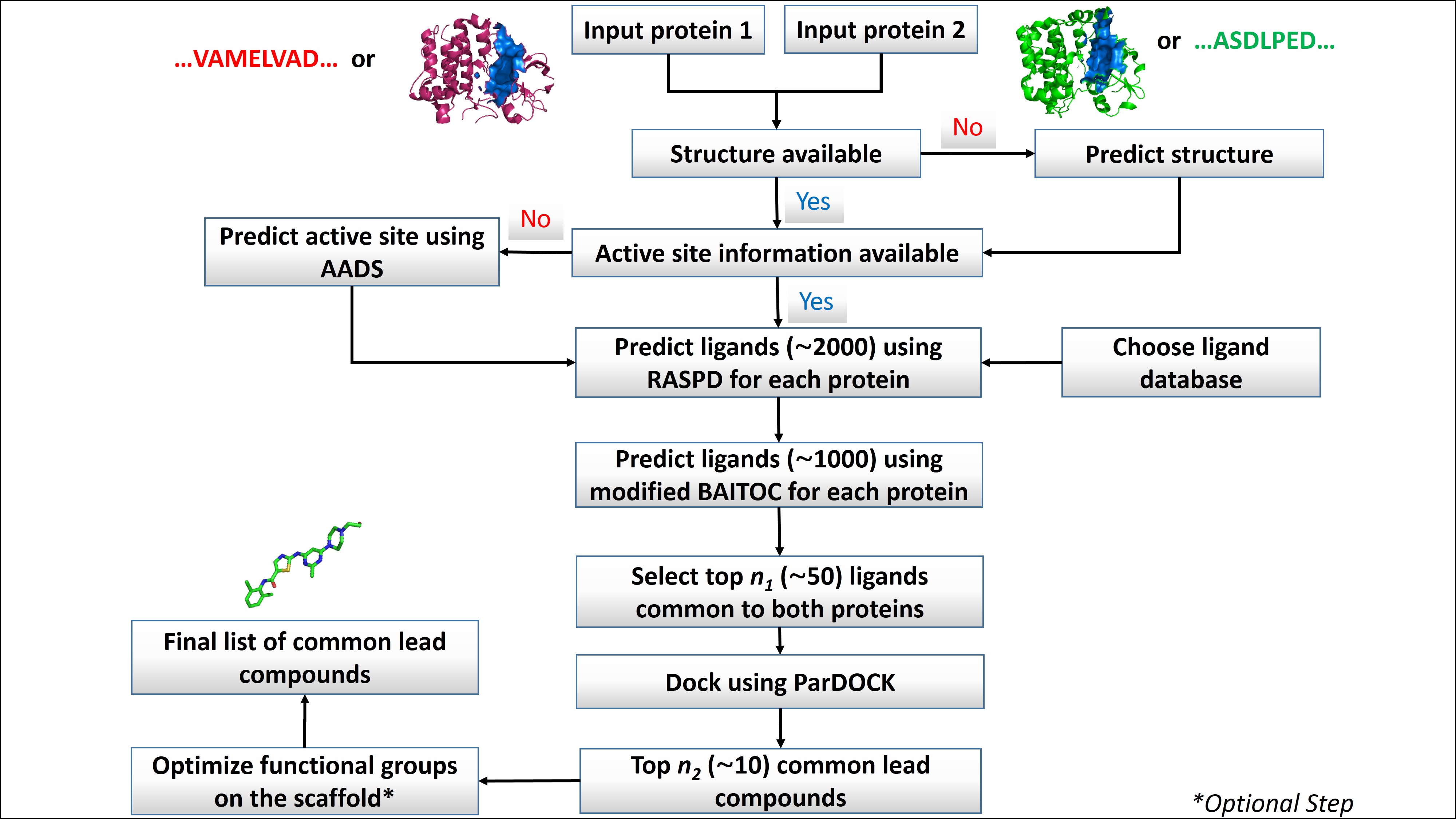
Multi Target Ligand Design (MTLD) is a web server that helps to identify common leads for any two protein targets. The basic principle of identifying common small molecules inhibiting multiple targets, remains the active site similarity and common interactive residues in their binding pockets. The protocol is based on the utilization of already established and validated softwares which are harnessed here to provide a set of common ligands for multiple proteins. A schematic representation of the methodology is shown below.
Step1: Input two protein targets (either sequence or structure)
Step2: If sequence is fed to system, it will generate tertiary structure for the input sequence provided.
Step3: After the structure generation it searches for most potent binding pocket using AADS program.
Step4: If structure is fed in step 1, it looks for binding pocket by identifying the ligands attached in the provided structure. In case of no ligand is found, it predicts binding pocket as in step3.
Step5: Both the proteins undergo virtual screening simultaneously against the user selected compound library. The output of this step is max 2000 hits.
Step6: Among the top 2000 molecules, hits are further scanned using modified BAITOC to filter best 1000 molecules.
Step7: The common hits identified among the top 1000s are then subjected to atomic level docking and scoring using ParDOCK.
Step8: The hits are then ranked on the basis of their binding free energies predicted with both the input protein targets.
Step9: User gets the result displayed as rank of all the hits identified along with their structure and best binding poses of each identified common hit with both the protein targets. In case of sequence submission user can also download the tertiary structure modelled for the given sequences from the result page. The list of used input parameters is also displayed on the result webpage.
Data will be deleted after 7 days from job completion.