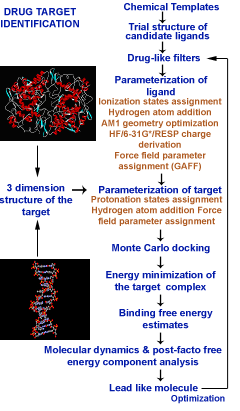
Sanjeevini software has been developed as a computational pathway paving the way expressely towards automating lead design, making any number of known or new candidate molecules out of a small but versatile set of building blocks called templates, screening them for drug likeness, optimizing their geomtery, determing partial atomic charges and assigning other force field parameters, docking the candidates in the active site of a given biological target , estimating the interaction/bidning energy, peforming molecular dynamics simulations with explicit solvent and salt on the biomolecular target, the candidate and the complex followed by a rigourous analysis of the binding free energy for further optimization. Presetly we have coupled Sanjeevini with AMBER and GAMESS for molecular mechanics and quantam mechanics calculations, respectively. There are total of six modules which makes Sanjeevini a complete drug design software. The source codes for all modules are written in FORTRAN, C and C++ computer languages with numerous interfacial UNIX based shell scripts which makes all the modules work like a pipeline such that output of the previous step becomes the input for the next step. The modules under Sanjeevini can also be used independent of the pathway.
|
To know more about Sanjeevini, refer to
Jain, T. and Jayaram, B. A computational protocol for predicting the binding affinities of zinc containing metalloprotein-ligand complexes. PROTEINS: Struct. Funct. Bioinfo., 2007, 67, 1167-1178.
Read Abstract
Shaikh S. A., Jain T., Sandhu G., Latha N. and Jayaram B., From Drug Target to Leads- Sketching, A Physicochemical Pathway for Lead Molecule Design In Silico ,Curr. Pharm. Des., 2007, 13, 34, 3454-3470.
Read Abstract
Shaikh S. A. and Jayaram B., A Swift All-Atom Energy Based Computational Protocol to Predict DNA ligand Binding Affinity and .Tm, J. Med.Chem. , 2007, 50, 2240-2244.
Read Abstract
Jain, T. and Jayaram, B. An all atom energy based computational protocol for predicting binding affinities of protein-ligand complexes. FEBS Letters. 2005, 579, 6659-6666.
Read Abstract
Latha, N., Jain, T., Sharma, P. and Jayaram, B. A free energy based computational pathway from chemical templates to lead compounds: a case study of COX-2 inhibitors. J. Biomol. Struct. Dyn. 2004, 21, 791-804.
Read Abstract
Jayaram, B., Latha, N.,Jain, T., Sharma, P., Gandhimathi, A., Pandey, V.S., Sanjeevini: A Comprehensive Active-Site Directed Lead Design Software. Indian Journal of Chemistry-A. 2006, 45A, 1834-1837. Read Full Text
Latha, N., and Jayaram, B. A Binding Affinity Based Computational Pathway for Active-Site Directed Lead Molecule Design: Some Promises and Perspectives. Drug Des. Rev.-Online. 2005, 2, 145-165.
Read Abstract
Kalra, P., Reddy, T.V. and Jayaram, B. Free energy component analysis for drug design: a case study of HIV-1 protease-inhibitor binding. J. Med. Chem. 2001, 44, 4325-4338.
Read Abstract
|