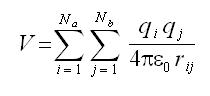
Molecular Dynamics is a form of computer simulation where atoms and molecules are allowed to interact for a period of time under known laws of physics, as general molecular system consist of large number of particles, it is impossible to find the properties of such complex system analytically. MD simulation circumvents this problem by using numerical methods. It represents an interface between laboratory experiments and theory and can be understood as virtual experiments. MD’s key contribution is creating awareness that molecules like proteins and DNA are machines. It provokes the relationship between molecular structure, movement and function. MD is a specialized discipline and computer simulation based on statistical mechanics. |
What is an Ensemble ? |
Electrostatic Interactions: The electrostatics interactions between pairs of point charges are calculated using coulomb’s law: |
|
Where and Na and Nb are the numbers of point charges in two molecules. The charge distribution can be represented as an arrangement of fractional point charges throughout the molecule. If charges are restricted to nuclear centers they are often refereed to as potential atomic charges or net atomic charges. |
Vanderwaals Interactions: It generally occurs at a short range and rapidly dies off as the interacting atoms move apart by a few Angstroms. This effect is modeled using equations in the following plot- |
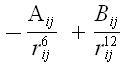 |
The ‘A’ & ‘B’ parameters control the depth and position (interatomic distance) of the potential energy well for a given pair of non bonded interacting atoms (e.g. 0: C, C:C, O:H etc) |
Energy Minimization: In molecular modeling we are interested in minimum points on the energy surface. Minimum energy arrangements of the atoms correspond to stable states of the system; any movement away from a minimum gives a configuration with a higher energy. There may be a very large number of minima on the energy surface. The minimum with the very lowest energy is known as the global energy minimum. |
Challenges in Molecular modeling: There is a difficulty of calculating free energies in silico i.e, free energies is usually expressed as the Helmholtz function A or the Gibbs function G. The Gibbs free energy is appropriate to a system with constant number of particles, temperature and pressure (constant NPT). |
Solvations(Continuum representations of the solvent): Most chemical processes takes place in a solvent so it is important to consider how the solvent affects the behavior of the system, in some cases solvent molecules are directly involved or in some cases solvent molecules are tightly bound. In other systems the solvent does not directly interact with the solute but it provide an environment that strongly affects the behavior of the solute. |
How Modelling is done ? Most reactions of interest does not take place in the gas phase,but in some medium be it in a solvent, in an enzyme or on the surface of a catalyst. The environment can have a significant impact upon the reaction by speeding it up or slowing it down or even changing the reaction pathway. The preferred technique for modeling chemical reaction is usually considered to be quantum mechanics. |