
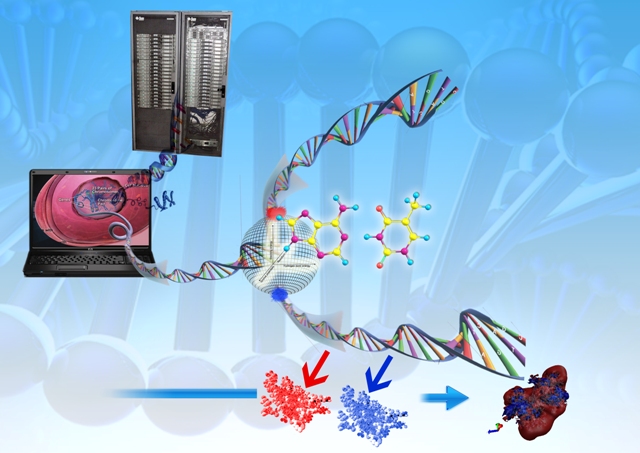

Design of a novel drug is one of the biggest challenges faced by the pharmaceutical industry. The use of computers accelerate the process of drug design which is a time intensive process, and also reduces the cost of whole process. Computational methods are used in various forms of drug discovery like QSAR, virtual screening and structure-based drug designing methods. Among these, structure based drug design is gaining importance due to rapid growth in structural data (available in RCSB & Nucleic acid Data Bank). This structural data can be used in molecular modeling to design lead molecules based on the structural features of the active site.
Research Initiave in the field of Drug Design @SCFBio
Pursuing the dream that once the gene target is identified and validated drug
discovery protocols could be automated using Bioinformatics & Computational Biology
tools. At IIT Delhi we have developed a computational protocol for active site directed
drug design. The suite of programs (christened “Sanjeevini”) has the potential to evaluate
and /or generate lead-like molecules for any biological target. The various modules of
this suite are designed to ensure reliability and generality. The software is currently being
optimized on Linux and Sun clusters for faster and better results.
Making a drug is more like designing an adaptable key for a dynamic lock. The Sanjeevini methodology consists of design of a library of templates, generation of candidate inhibitors, screening candidates via drug-like filters, parameter derivation via quantum mechanical calculations for energy evaluations, Monte Carlo docking and binding affinity estimates based on post facto analyses of all atom molecular dynamics trajectories. The protocols tested on Cyclooxygenase-2 as a target could sucessfully distinguish NSAIDs (Nonsteroidal Anti-inflammatory drugs) from non-drugs. Validation on other targets is in progress.
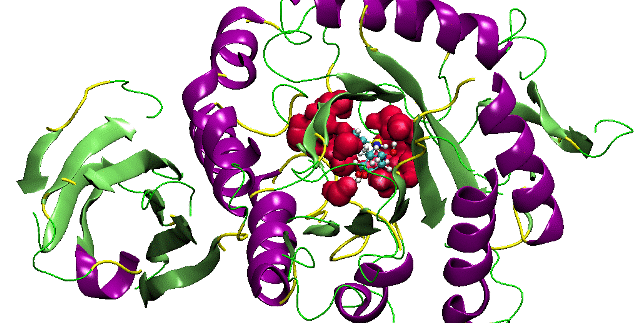
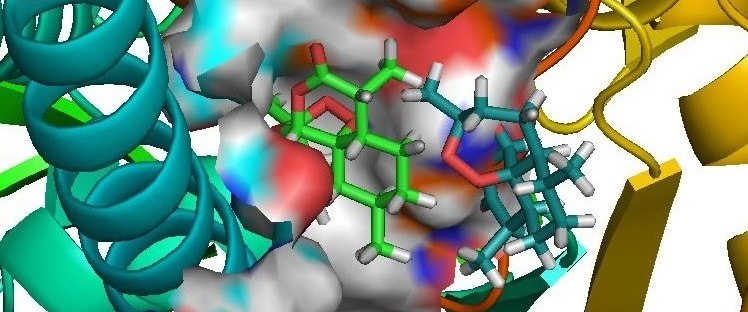
Sanjeevini (Receptor Ligand Interaction)
A comprehensive active site directed lead compound design software, based on the on-going research in our laboratory. The computational pathway integrates several protocols proceeding from the design of chemical templates to lead-like molecules, given the three dimensional structure of the target protein and a definition of its active site. A conscious attempt has been made to handle the target biomolecule and the candidate drug molecules at the atomic level retaining system independence while providing access for systematic improvements at the force field level. Concerns related to geometry of the molecules, partial atomic charges, docking of candidates in the active site, flexibility and solvent effects are accounted for at the current state-of-the-art. To ensure theoretical rigor, binding free energy estimates are developed for candidate molecules with the target protein within the framework of statistical mechanics.
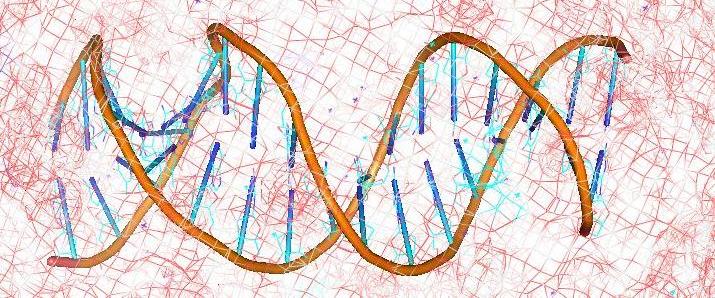
DNA Drug Interaction
DNA recognition involves a complex interplay of a variety of interactive forces. Hydrogen bonding interactions, strong electrostatic interactions arising due to phosphates, mobile counter-ions and hydration, van der Waals contacts, the energetic consequences of sequence dependent structural/conformational adaptation of DNA and hydrophobic interactions play critical roles in constructing the energy profile of the binding process. Thus, developing a molecular view of the thermodynamics of DNA recognition is essential to the design of DNA binders.